Введение
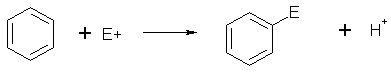
Электрофильное замещение, несомненно, составляет самую важную группу реакций ароматических соединений. Вряд ли найдется какой-нибудь другой класс реакций, который так детально, глубоко и всесторонне исследован как с точки зрения механизма, так и с точки зрения применения в органическом синтезе. Именно в области электрофильного ароматического замещения впервые была поставлена проблема связи между структурой и реакционной способностью, которая является основным предметом изучения в физической органической химии. В общем виде этот тип реакций ароматических соединений может быть представлен следующим образом:
ArH + E+  ArE + H+
ArE + H+
1. Литературный обзор
1.1 Электрофильное замещение в ароматическом ряду
Эти реакции характерны не только для самого бензола, но и вообще для бензельного кольца, где бы оно ни находилось, а также для других ароматических циклов — бензоидных и небензоидных. Реакции электрофильного замещения охватывают широкий круг реакций: нитрование, галогенирование, сульфирование и реакции Фриделя — Крафтса свойственны почти всем ароматическим соединениям; реакции типа нитрозирования и азосочетания присущи лишь системам с повышенной активностью; такие реакции, как десульфирование, изотопный обмен, и многочисленные реакции циклизации, которые с первого взгляда кажутся совсем различными, но которые также оказывается целесообразным отнести к реакциям того же типа.
1.1.1 Электрофильные агенты
Электрофильные агенты Е+ , хотя наличие заряда не обязательно, т.к. электрофил может быть и незаряженной электронодефицитной частицей (например, SO3, Hg(OCOCH3)2 и т.п.). Условно их можно разделить на три группы: сильные, средней силы и слабые.
Сильные электрофилы
NO2+(Ион нитрония, нитроил-катион); комплексы Cl2 или Br2 с различными кислотами Льюиса (FeCl3, AlBr3, AlCl3, SbCl5 и т.д.); H2OCl + , H2OBr + , RSO2+ , HSO3+ , H2S2O7 . Сильные электропилы взаимодействуют с соединениями ряда бензола, содержащими как электронодонорные, так и практически любые электроноакцепторные заместители.
Электрофилы средней силы
Комплексы алкилгалогенидов или ацилгалогенидов с кислотами Льюиса (RCl. AlCl3, RBr. GaBr3, RCOCl. AlCl3 и др.); комплексы спиртов с сильными кислотами Льюиса и Бренстеда (ROH. BF3, ROH. H3PO4, ROH. HF). Реагируют с бензолом и его производными, содержащими электронодонорные (активирующие) заместители или атомы галогенов (слабые дезактивирующие заместители), но обычно не реагируют с производными бензола, содержащими сильные дезактивирующие электроноакцепторные заместители (NO2, SO3H, COR,CN и др.).
Слабые электрофилы
Катионы диазония ArN+є N, иминия CH2=N+ H2, нитрозония NO+(нитрозоил-катион); оксид углерода (IY) СО2 (один из самых слабых электрофилов). слабые электрофилы взаимодействуют только с производными бензола, содержащими очень сильные электронодонорные заместители (+М)-типа (OH, OR, NH2, NR2, O- и др.).
1.1.2 Механизм электрофильного ароматического замещения
В настоящее время ароматическое электрофильное замещение рассматривается как двухстадийная реакция присоединения-отщепления с промежуточным образованием аренониевого иона, называемого σ-комплексом
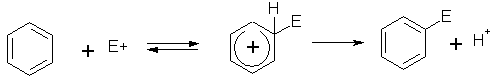
I-Аренониевый ион (  -комплекс), как правило, короткоживущий. Такой механизм получил название SEAr, т.е. SЕ (аренониевый). В этом случае на первой стадии в результате атаки электрофила циклическая ароматическая 6-электронная π-система бензола исчезает и заменяется в интермедиате I на нециклическую 4-электронную сопряженную систему циклогексадиенильного катиона. На второй стадии вновь восстанавливается ароматическая
-комплекс), как правило, короткоживущий. Такой механизм получил название SEAr, т.е. SЕ (аренониевый). В этом случае на первой стадии в результате атаки электрофила циклическая ароматическая 6-электронная π-система бензола исчезает и заменяется в интермедиате I на нециклическую 4-электронную сопряженную систему циклогексадиенильного катиона. На второй стадии вновь восстанавливается ароматическая  -система за счет отщепления протона.Строение аренониевого иона I изображают различными способами:
-система за счет отщепления протона.Строение аренониевого иона I изображают различными способами:
$IMAGE6$ $IMAGE7$
Наиболее часто употребляется первая формула. σ-комплекс будет гораздо лучше стабилизироваться донорными заместителями, находящимися в орто- и пара-положениях, чем донорными заместителями в мета-положении.
π-Комплексы
Как известно, арены являются π-основаниями и могут образовывать донорно-акцепторные комплексы со многими электрофильными реагентами.Так, при пропускании сухих газообразных HCl или DCl в раствор бензола, толуола, ксилолов или других полиалкилбензолов в н-гептане при -78оС удалось обнаружить образование молекулярных комплексов состава 1:1 (Г.Браун, 1952 г.).
Эти комплексы не окрашены, их растворы в ароматических углеводородах неэлектропроводны. При растворении газообразного DCl в бензоле, толуоле, ксилолах, мезитилене, пентаметилбензоле не происходит обмен H на D. Поскольку растворы комплексов не проводят электрический ток, они не являются ионными частицами, т.е. это не аренониевые ионы.
Такие донорно-акцепторные комплексы получили название π-комплексов. Например, кристаллы комплексов бензола с бромом или хлором состава 1:1 согласно рентгеноструктурным данным состоят из цепочек чередующихся молекул π-донора состава (C6H6) и акцептора (Cl2,Br2), в которых молекула галогена расположена перпендикулярно плоскости кольца вдоль оси, проходящей через его центр симметрии.
σ-комплексы (аренониевые ионы)
При введении в раствор HCl и DCl в алкилбензолах AlCl3 или AlBr3 раствор начинает проводить электрический ток. Такие растворы окрашены и их окраска изменяется от желтой до оранжево-красной при переходе от пара-ксилола к пентаметилбензолу. В системах ArH-DCl-AlCl3 или ArH-DF-BF3 атомы водорода ароматического кольца уже обмениваются на дейтерий. Электропроводность растворов определенно указывает на образование ионов в тройной системе арен-галогенводород-галогенид алюминия. Строение таких ионов было установлено с помощью ЯМР-спектроскопии на ядрах 1Н и 13С в системе ArH-HF(жидк)-BF3 или ArH-HF-SbF5 в SO2ClF при низкой температуре.
1.1.3 Классификация заместителей
Монозамещенные бензолы С6Н5Х могут быть более или менее реакционноспособны, чем сам бензол. Если в реакцию ввести эквивалентную смесь С6Н5Х и С6Н6, то замещение будет происходить селективно: в первом случае в реакцию будет вступать преимущественно С6Н5Х, а во втором случае - преимущественно бензол.
В настоящее время заместители делят на три группы с учетом их активирующего или дезактивирующего влияния, а также ориентации замещения в бензольном кольце.
1. Активирующие орто-пара-ориентирующие группы. К ним относятся: NH2, NHR, NR2, NHAc, OH, OR, OAc, Alk и др.
2. Дезактивирующие орто-пара-ориентирующие группы. Это галогены F, Cl, Br и I.
3. Дезактивирующие мета-ориентирующие группы. Эту группу составляют NO2, NO, SO3H, SO2R, SOR, C(O)R, COOH, COOR, CN, NR3+ ,CCl3 и др. Это ориентанты II-го рода.
Естественно, что существуют и группировки атомов промежуточного характера, обусловливающие смешанную ориентацию. К ним, например, относятся: CH2NO, CH2COCH3, CH2F, CHCl2, CH2NO2, CH2CH2NO2, CH2CH2NR3+, CH2PR3+, CH2SR2+ и др.
1.2 Электрофильное замещение в π-избыточных гетероциклах
Фуран, пиррол и тиофен обладают значительной реакционной способностью по отношению к обычным электрофильным реагентам. В этом смысле они напоминают наиболее реакционно-способные производные бензола, такие, как фенолы и анилины. Повышенная чувствительность к электрофильному замещению вызвана несимметричным распределением заряда в этих гетероциклах, в результате чего на углеродных атомах цикла имеется больший отрицательный заряд, чем в бензоле. Фуран обладает несколько большей реакционной способностью, чем пиррол, а наименее реакционноспособен тиофен.
1.2.1 Электрофильное замещение пиррола
В то время как пиррол и его производные не склонны к реакциям нуклеофильного присоединения и замещения, они очень чувствительны к электрофильным реагентам, и реакции пирролов с такими реагентами протекают практически исключительно как реакции замещения. Незамещенный пиррол, N- и С-моноалкилпирролы и в наименьшей степени С,С-диалкилпроизводные полимеризуются в сильнокислых средах, поэтому большинство электрофильных реагентов, использующихся в случае производных бензола, не применимы для пиррола и его алкилпроизводных.
Однако при наличии в пиррольном цикле электроноакцепторных групп, препятствующих полимеризации, например, таких, как сложноэфирная, становится возможным использование сильнокислых сред, нитрующих и сульфирующих агентов.
Протонирование
В растворе наблюдается обратимое присоединение протона по всем положениям пиррольного цикла. Наиболее быстро протонируется атом азота, присоединение протона по положению 2 проходит в два раза быстрее, чем по положению 3. В газовой фазе при использовании кислот умеренной силы, таких, как C4H9+ и NH4+, пиррол протонируется исключительно по атомам углерода, причем склонность к присоединению протона по положению 2 выше, чем по положению 3. Наиболее термодинамически стабильный катион - 2Н-пирролиевый ион - образуется при присоединении протона по положению 2 и определяемое значение рКа для пиррола связано именно с этим катионом. Слабая N-основность пиррола обусловлена отсутствием возможности для мезомерной делокализации положительного заряда в 1H-пирролиевом катионе.
$IMAGE8$
Значение рКа определено для большого числа производных пиррола, а сам незамещенный пиррол — чрезвычайно слабое основание со значением рКа -3,8. Основность пиррольного цикла весьма быстро увеличивается при введении алкильных заместителей, и для 2,3,4,5-тетраметилпиррола рКа равен +3,7, что соответствует полному протонированию всех молекул пиррола в вышеприведенных условиях (для сравнения рКа анилина +4,6). Таким образом, алкильные группы оказывают необычайное стабилизирующее влияние на катионы - пирролы, содержащие трет-бутильные группы, при протонировании образуют стабильные кристаллические соли.
$IMAGE9$ $IMAGE10$
Реакции протонированных пирролов
2Н- и ЗН-Пирролиевые катионы в сущности представляют собой иминиевые ионы и, следовательно, обладают свойствами электрофилов. Эти катионы играют ключевую роль в процессах полимеризации и восстановления пирролов в присутствии кислот. При взаимодействии пирролов с гидрохлоридом гидроксиламина, сопровождающимся раскрытием цикла и образованием 1,4-диоксимов, вероятно, образуется более реакционноспособный ЗН-пирролиевый катион. Для защиты аминогруппы первичных аминов могут быть превращены в 1-К-2,5-диметилпирролы, затем защитная группа может быть удалена с помощью описанной выше реакции с гидроксиламином.
$IMAGE11$
Нитрование
Нитрующую смесь, применяемую для нитрования производных бензола, нельзя использовать в случае пиррола, поскольку это приводит к его полному разложению. Однако нитрование пиррола возможно при использовании ацетилнитрата при низких температурах, причем преимущественно образуется 2-нитро-пиррол. Ацетилнитрат получают при смешивании дымящей азотной кислоты с уксусным ангидридом, и в результате образуется уксусная кислота и достигается удаление сильной минеральной кислоты. При нитровании пиррола с использованием ацетилнитрата активность положения 2 в 1,3 • 105, а положения 3 в 3 • 104 раза выше активности бензола.
$IMAGE12$
Введение заместителя к атому азота увеличивает долю продукта нитрования по положению 3 в смеси продуктов реакции: так, введение метального заместителя обусловливает получение смеси продуктов β- и α-нитрования в соотношении 1:3. Более объемная трет-бутильная группа приводит даже к обращению относительной реакционной способности — продукты β- и α-нитрования образуются в соотношении 4:1. Полного подавления реакции нитрования по α-положению пиррола можно достигнуть при введение к атому азота триизопропилсилильной (TIPS) группы; использование последней чрезвычайно важно при синтезе 3-производных, так как в последствии она может быть легко удалена.
$IMAGE13$
Сульфирование и реакции с использованием других серосодержащих электрофильных реагентов
Для сульфирования пирролов используются мягкие сульфирующие агенты, не обладающие свойствами кислоты; так, комплекс пиридин — триоксид серы мягко превращает пиррол в пиррол-2-сульфонат .
$IMAGE14$
Реакции с использованием других серосодержащих эл